Research
Our research focuses on the genomic basis of contemporary evolution in natural populations. We want to understand the contributions of different evolutionary forces in shaping patterns of variation across the genome through space and time, and to link genetic variation to variation in individual phenotypes, fitness, and eventually population dynamics. We answer these questions by developing genomic resources and statistical tools that combine evolutionary genomics and pedigree data from long-term demographic studies of vertebrates such as the Florida Scrub-Jay.
Short-term evolutionary dynamics
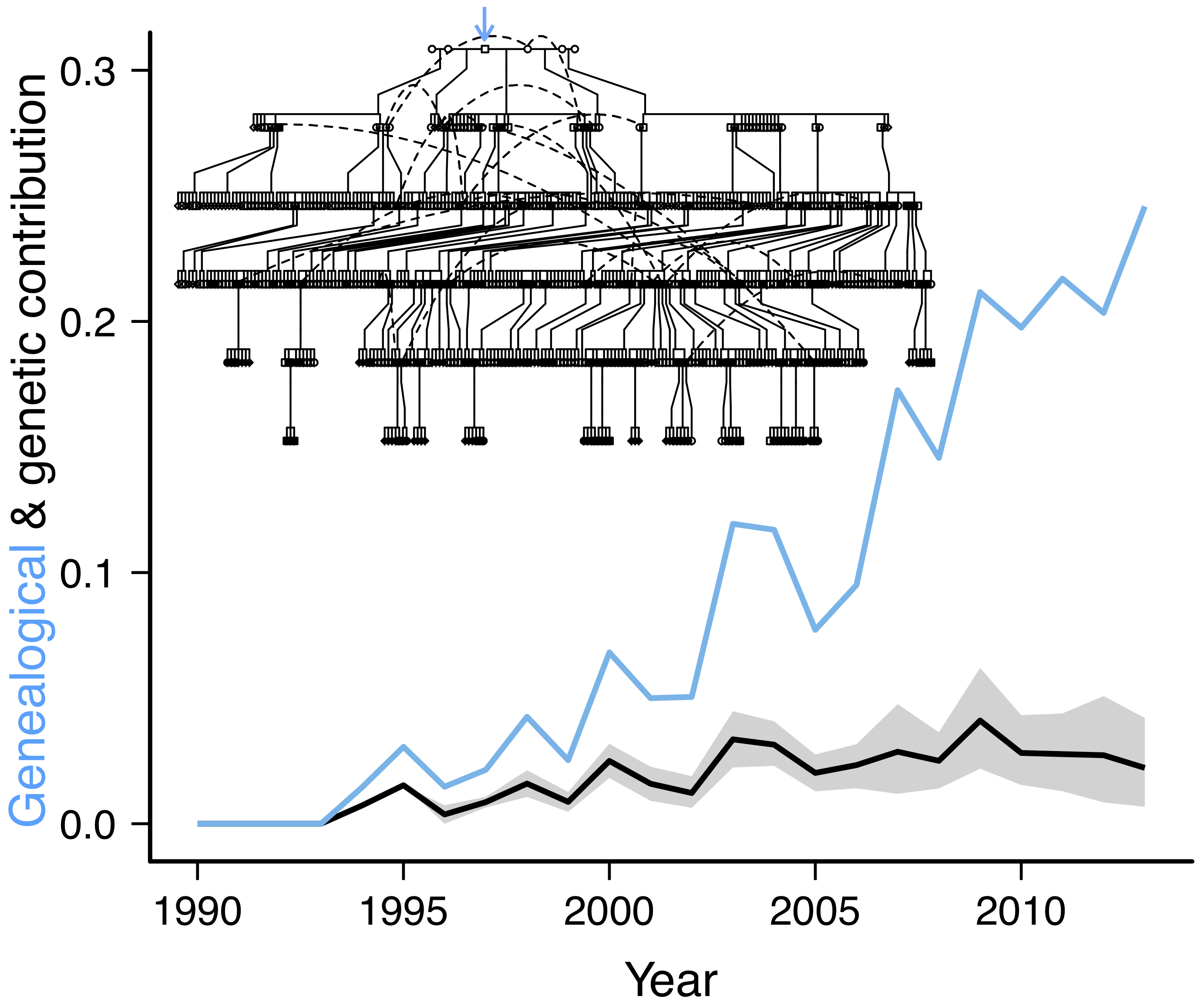
Population geneticists typically think about evolutionary processes such as genetic drift, selection, and gene flow in terms of allele frequency change over time. But allele frequencies change because of individual variation in survival, reproduction, and movement, the mechanisms underlying different individual genetic contributions to the population over time. Therefore the population pedigree, the relationships among all individuals in a population over time, allows direct observation of the processes underlying allele frequency change in natural populations. We combine genomic data with extensive pedigree information to illustrate the evolutionary mechanisms shaping patterns of genetic variation through time and space. For instance, we empirically demonstrated how limited dispersal can generate a pattern of isolation-by-distance over short spatial scales in just a few generations. Recent work empirically linked expected individual genetic contributions to individual fitness and predictions of allele frequency change, and quantified the relative roles of evolutionary processes governing allele frequency change. We are now incorporating recombination and tracking the inheritance of the entire genome down the pedigree to determine how individual variation in lifetime reproductive success results in changes in population genetic variation and identify regions of the genome associated with fitness. Our work provides a deeper insight into the process of evolution at the genomic level.
Maintenance of variation in fitness-related traits
One of the deepest unresolved paradoxes in evolutionary biology is the persistence of high levels of genetic variation in the face of selection. Why do we observe abundant genetic variation in many traits associated with fitness in natural populations? We are assessing the role of fluctuating selection, antagonistic pleiotropy, and sexual conflict in maintaining variation in the Florida Scrub-Jay. To tackle this question, we are using gene dropping simulations to test for selection across generations, and selection component analysis to test for selection on specific life-cycle stages within generations. We are extending our tests of selection to detect signals of polygenic adaptation. Applying these integrative methods to one of few natural systems with accurate measures of individual fitness provides a powerful framework for measuring short-term selection and evaluating the role of selection in maintaining variation at fitness-related traits in nature.
Genomics of population decline

Despite much theoretical work describing the genetic consequences of population decline, few thorough empirical validations of these theories have been done at the genome-wide level in wild populations. The Florida Scrub-Jay is one of the longest-studied endangered species in the world and an unparalleled model system for conservation genomics. We use longitudinal studies to assess the genomic and fitness consequences of habitat fragmentation. Previous work investigated temporal variation in gene flow, inbreeding, and fitness and showed that, contrary to conventional wisdom, small peripheral populations may play a vital role in maintaining genetic variation in larger and seemingly stable populations. We continue to study questions concerning the genetic basis of inbreeding depression, the long-term contributions of immigrants to population genetic diversity and fitness, and the role of population size and isolation in predicting population viability. This work will increase our understanding of the population genetic consequences of declining population size and inform conservation strategies for the Florida Scrub-Jay and other declining species.
Methods development
We develop methods to address limitations in current genomic and population genetic approaches. For example, the female-specific W chromosomes and male-specific Y chromosomes remain difficult to assemble using whole-genome shotgun methods. We developed a novel method for identifying sequences specific to the heterogametic sex chromosome, which has facilitated future genome assembly efforts and enhanced studies of sex chromosome evolution. To improve the SNP discovery process in non-model organisms, we developed a statistical framework for filtering spurious variant calls and identifying putative sex-linked sites using Mendelian inheritance patterns in nuclear families. Much of our current work focuses on developing population genetic inference methods that incorporate pedigree information. These tools are broadly applicable both within our own research program and to other groups involved in ecological and evolutionary genetics research.